Introduction
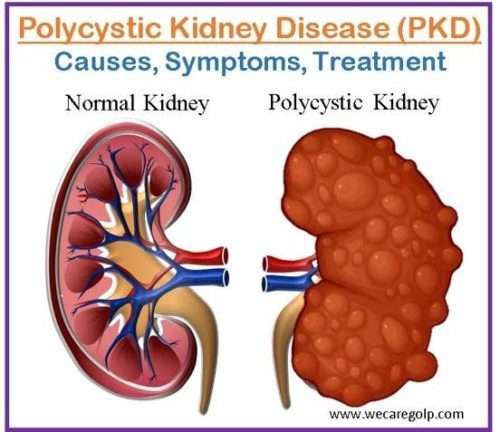
Polycystic kidney disease (PKD) is a genetic condition characterized by bilateral renal cysts (fluid-filled sacs) and renal enlargement. It is a progressive multisystem illness involving other organs e.g., liver, pancreas, spleen, etc. Autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive polycystic kidney disease (ARPKD) are two primary types of PKD.
Kidney failure can develop from PKD cysts, which can significantly expand the kidneys while replacing most of their normal structure. The patient needs dialysis (hemodialysis or peritoneal dialysis) or a kidney transplant when PKD causes renal failure, which often occurs after several years. With the most prevalent form of PKD, around 50% of patients develop kidney failure, often known as end-stage renal disease.
Cysts in the liver and issues with other organs, such as the blood arteries in the brain and heart, can also be brought on by PKD. Doctors can identify PKD from the often innocuous “simple” cysts that frequently occur in the kidneys in later life by counting the number of cysts and the difficulties they cause. Cysts that form in PKD can affect kidney function by compromising the organ’s capacity to filter waste and control fluid and electrolyte balance. This can cause a variety of symptoms and consequences, such as high blood pressure, chronic kidney disease (CKD), kidney stones, urinary tract infections (UTIs), and, in extreme situations, kidney failure.
Incidence
- It is the most common hereditary cause of renal failure in adults, accounting for 6% to 8% of dialysis patients in the United States.
- By the age of 60, half of PKD patients require renal replacement therapy (RRT).
- Cysts can be seen in utero or children, although clinical signs do not show until the third or fourth decade of life in most of cases.
- Worldwide, ADPKD affects roughly 4 to 7 million people, accounting for 7-15% of patients on RRT.
- Males have somewhat worse ADPKD than females, although the difference is not statistically significant.
- Symptoms usually worsen with aging. Children with ADPKD seldom manifest with renal failure.
Classification of Polycystic Kidney Disease
Autosomal dominant polycystic kidney disease (ADPKD)
The most frequent hereditary type of PKD is ADPKD. The onset of symptoms typically occurs between the ages of 30 and 40, but they can occur earlier, even in childhood or utero. About 90% of all PKD cases involve ADPKD. Cyst formation and kidney and other organ enlargement are hallmarks of this multisystemic, progressive disease. Other organs affected include the liver, pancreas, and spleen. By the age of 60, up to 50% of people with ADPKD require renal replacement therapy (RRT).
Autosomal recessive polycystic kidney disease (ARPKD)
It is an uncommon hereditary type of disease. ARPKD symptoms appear in the first months of life, even in the womb. ARPKD is characterized by cystic dilatation of the renal collecting ducts, as well as various degrees of hepatic abnormalities such as biliary dysgenesis and periportal fibrosis. Although 50–60% of newborns may not show signs of hepatic involvement, infants and toddlers are regularly found to have the illness.
Causes of Polycystic Kidney Disease
ADPKD
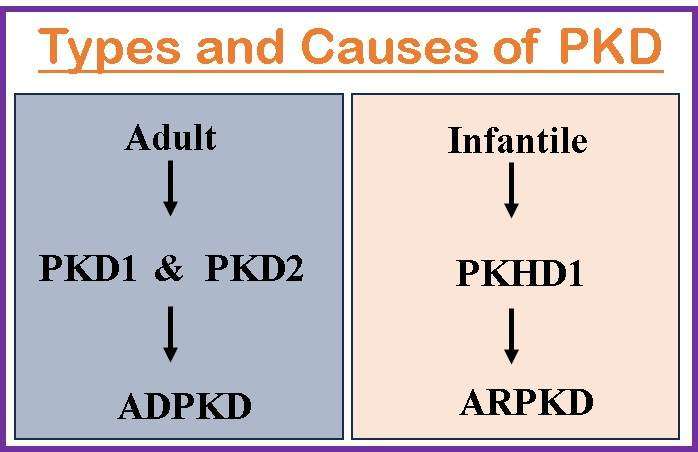
It is primarily caused by genetic mutations, either PKD1 or PKD2. These genes code for proteins that are important in the development and maintenance of the kidneys. ADPKD is inherited in an autosomal dominant manner, which means that an afflicted person has a 50% probability of transferring the mutant gene to each of their offspring. The faulty proteins disturb the normal structure and function of kidney cells, resulting in cyst development. Two genes at least are involved with ADPKD.
- Most occurrences of ADPKD are caused by PKD1, which is situated on 16p13.3. Polycystin 1 (a protein of 4304 amino acids) is encoded by PKD1, is involved in intracellular calcium transport and cell cycle control, and interacts with polycystin 2.
- 15% of instances of ADPKD are PKD2 patients. PKD2 codes for polycystin 2, which resembles polycystin 1 structurally. It belongs to the group of calcium channels that are activated by voltage.
The epithelial cells of the renal tubules and other parts of the renal cell epithelium are home to polycystins 1 and 2. Both are present in the main cilium of kidney epithelial cells and form heteromeric complexes there. A mechanical receptor, the main cilium is thought to be able to detect variations in tubular fluid flow and convert them into intracellular calcium signaling. Compared to ADPKD2, ADPKD1 is more severe.
ARPKD
It is a less common variant of PKD that usually appears in infancy or early childhood. Mutations in the PKHD1 (polycystic kidney and hepatic disease 1) gene, which codes for the protein fibrocystin, cause it. The absence of functioning fibrocystin causes aberrant growth and dilatation of the renal tubules, which culminates in cyst development. Affected individuals must inherit two copies of the defective gene, one from each parent, to develop ARPKD.
Signs and Symptoms of Polycystic Kidney Disease
ADPKD
Renal manifestations
- Bilateral enlargement of the kidneys
- Flank pain or abdominal discomfort caused by cyst expansion or bleeding
- Hematuria due to cyst rupture or bleeding
- Hypertension is often early onset and difficult to control
- Renal insufficiency or progressive decline in kidney function
- Recurrent UTIs
- Renal stones or urinary obstruction due to cyst compression on the urinary tract
- Microalbuminuria, proteinuria
Extrarenal manifestations
- Cysts in other organs such as the liver, pancreas, spleen, and seminal vesicles
- Intracranial aneurysms can lead to strokes or other neurological complications
- Cardiac valve abnormalities
- Diverticulosis which can lead to diverticulitis
- Hernias due to weakened connective tissue
ARPKD
Pediatric manifestations
- Enlarged, palpable kidneys in infants
- Pulmonary hypoplasia due to compression by enlarged kidneys
- Respiratory distress shortly after birth
- Hypertension, often severe in infants
Childhood/adolescent manifestations
- Renal manifestations similar to ADPKD, including kidney enlargement, renal insufficiency, and hypertension
- Hepatomegaly due to cysts in the liver
- Portal hypertension (increased pressure in the portal vein system) due to liver involvement
- Biliary tract abnormalities and cholangitis
- Growth retardation and poor weight gain
- Varicocele in males
Pathophysiology of Polycystic Kidney Disease
Specific genetic mutations are responsible for PKD. Mutations in the PKD1 or PKD2 genes cause ADPKD by interfering with the generation or function of proteins important in kidney growth and maintenance. Mutations in the PKHD1 gene cause the generation of aberrant or non-functional fibrocystin proteins in ARPKD. Genetic mutations cause renal tubular epithelial cell dysfunction, which lines the kidney tubules. These cells are critical in maintaining the kidneys’ proper shape and function. Dysfunctional renal tubular epithelial cells proliferate excessively, meaning they divide and multiply.
Cysts are formed as a result of aberrant cell growth. The afflicted regions’ renal tubular epithelial cells leak fluid into the cysts. This fluid collection causes cysts to expand and, as a result, healthy renal tissue to be compressed. Because the defective renal tubular epithelial cells continue to secrete fluid, the cysts expand and increase with time. The cysts can develop in different areas of the kidneys and range in size and number.
Cysts compress and displace normal kidney tissue, including nephrons, which are the functional components of the kidneys responsible for filtering blood and generating urine, as they expand. As a result of the compression and displacement, normal kidney function gradually declines. The ongoing growth and expansion of cysts, as well as the compression of surrounding tissue, cause an inflammatory response and connective tissue deposition (fibrosis) in the kidney interstitial space. Interstitial fibrosis affects normal kidney architecture and inhibits renal function further.
As PKD advances, the cumulative effects of cyst development, compression, displacement, and interstitial fibrosis reduce renal function. The kidneys lose their ability to filter waste and maintain fluid and electrolyte balance. This malfunction can lead to a variety of problems, including hypertension, renal insufficiency, kidney failure, and other systemic consequences.
Diagnosis of Polycystic Kidney Disease
Physical examination
- Enlarged kidney
- Enlarged liver
- Heart murmur
- High blood pressure
- Pain
Blood tests
- Renal function test
- Liver function test
- Complete blood cell count-Hematocrit level is increased
- Intact parathyroid hormone assay
Urine tests
- Urine culture (Escherichia coli, Klebsiella and Proteus species, and other Enterobacteriaceae)
- Uric acid determination
- Microalbuminuria
- Hematuria
- Proteinuria
Genetic testing
- ADPKD and ADPLD may all be distinguished via genetic testing.
- The presence of either PKD1 or PKD2 subtypes in ADPKD alters the prognosis and may aid in treatment.
Imaging
- In the workup of individuals with autosomal dominant polycystic kidney disease, ultrasonography (USG) is the method of choice. It is also great for screening patients’ relatives.
- If USG cannot detect small cysts, a computed tomography (CT) scan, magnetic resonance imaging (MRI), and magnetic resonance angiography (MRA) might be helpful.
Treatment of Polycystic Kidney Disease
Goals for management are:
- Maintain blood pressure
- Treat UTIs
- Treatment for hematuria
- Relieve abdominal discomfort caused by enlarged kidneys
- Treat the hemorrhage
Treat UTIs
UTIs affect 30-50% of ADPKD patients, with women being the most affected. The most frequent infections are Gram-negative bacteria. It is critical to distinguish between infections of the bladder, renal parenchyma, and cysts since therapy for each illness differs.
Antibiotics that are mostly used to treat UTIs are.
- Ciprofloxacin
- Trimethoprim-sulfamethoxazole
- Clindamycin
- Chloramphenicol
Control blood pressure
The preferred antihypertensive drugs in ADPKD are:
- Angiotensin-converting enzyme (ACE) inhibitors
- Angiotensin receptor blockers (ARBs)
Control abnormalities related to renal failure
- Potassium citrate is the preferred therapy for stone-forming problems such as
- Uric acid stones
- Hypocitraturic calcium oxalate stones
- Distal acidification disorders
- Surgical cyst decompression is used to treat the condition, which relieves discomfort in 60-80% of patients with abdominal pain due to enlarged kidneys.
- Kidney failure necessitates the use of pharmaceuticals to maintain electrolyte levels for example:
- Phosphate binder (calcium carbonate, calcium acetate, sevelamer, lanthanum carbonate)
- Calcitriol
- Diuretics
- Blood pressure medications
- For optimum blood pressure management, approximately 62% of patients with renal insufficiency require at least two antihypertensive medications.
- Nonsteroidal anti-inflammatory medicines (NSAIDs) should be avoided since they can impair renal function and exacerbate hyperkalemia.
Treat hemorrhage and hematuria
Hematuria is common in ADPKD patients and typically self-limiting. It is frequently caused by a cyst rupture or stone passage.
- Cyst hemorrhage can deal with conservative therapy to prevent clots from obstructing blood vessels which includes
- Bed rest
- Painkillers
- Increased fluid intake
- The bleeding can occasionally be significant enough to cause hemodynamic instability, necessitating
- Hospitalization and
- Blood transfusions
- Cyst infections necessitate the use of gyrase inhibitor medications such as
- Ciprofloxacin
- Chloramphenicol
- Clindamycin
- Trimethoprim-sulfamethoxazole
At later stage
Along with the above-mentioned therapy, the following treatment may be needed.
- Dialysis (hemodialysis or peritoneal dialysis)
- Kidney transplant
- Liver transplant
Complications of Polycystic Kidney Disease
- Chronic kidney disease (CKD)
- End-stage renal disease (ESRD)
- Renal stones
- Hypertension (HTN)
- Valvular heart disease
- Polycystic liver disease
- Liver and pancreatic cysts
- Cerebral aneurysms
- UTIs (cystitis, acute pyelonephritis), cyst infection, perinephric abscesses
Prevention of Polycystic Kidney Disease
PKD is not a preventable disease. However, the progression can be slowed down with the following interventions and help reduce the complications like kidney failure.
- Consult a nephrologist as soon as possible if you have a family history of PKD to avoid risk
- Prevent and manage diabetes and high blood pressure
- Compliance with the medicines as prescribed
- Adopt the healthy lifestyle
- Do regular exercise (at least 30 mins per day)
- Reduce stress
- Maintain a healthy weight
- Control the blood pressure (less than 120/80 mmHg)
- Avoid smoking
- Drink less alcohol
- Sleep seven to eight hours each night
- Follow renal diet
- Avoid contact sports like hockey, football, etc. to avoid cysts bursting due to trauma
Prognosis
- The prognosis of polycystic kidney disease varies based on several factors. In general, the long-term outlook for people with PKD is marked by slow development of the disease. However, the pace of development and severity of consequences might vary greatly amongst patients.
- The prognosis of ADPKD can range from mild to severe. Some people may experience delayed disease development with little or no symptoms throughout their lives.
- Kidney failure is a major issue in people with PKD. A fraction of people with ADPKD will eventually develop ESRD, in which the kidneys can no longer function properly.
- The prognosis for ARPKD, which is often diagnosed in infancy or youth, can be worse. ARPKD frequently results in more widespread kidney involvement and consequences, such as reduced kidney function and liver issues. ARPKD prognosis varies greatly depending on disease severity and the availability of effective medical therapies.
Summary
Polycystic kidney disease (PKD) is an inherited condition that causes numerous cysts to form in the kidneys. ADPKD and ARPKD are the two most common types. ADPKD, which often appears in adulthood, has a varied prognosis. While some people have delayed disease development and few symptoms, others might develop consequences including high blood pressure, renal failure, and cysts in other organs. ARPKD diagnosed in children or infancy has a worse prognosis, with substantial kidney and liver damage.
Genetic factors, lifestyle decisions, and concurrent medical disorders can all impact the rate of development. Early identification, frequent monitoring, blood pressure control, and a healthy lifestyle, on the other hand, can help reduce disease development and improve outcomes. Individual prognoses may differ, thus seeking tailored advice from healthcare providers acquainted with PKD is vital.
References
- Bergmann, C., Guay-Woodford, L. M., Harris, P. C., Horie, S., Peters, D. J., & Torres, V. E. (2018). Polycystic kidney disease. Nature reviews Disease primers, 4(1), 50. https://www.nature.com/articles/s41572-018-0047-y
- Ghata, J., & Cowley Jr, B. D. (2011). Polycystic kidney disease. Comprehensive Physiology, 7(3), 945-975. https://doi.org/10.1002/cphy.c160018
- Harris, P. C., & Torres, V. E. (2009). Polycystic kidney disease. Annual review of medicine, 60, 321-337. https://doi.org/10.1146/annurev.med.60.101707.125712
- Igarashi, P., & Somlo, S. (2007). Polycystic kidney disease. Journal of the American Society of Nephrology, 18(5), 1371-1373. Doi:10.1681/ASN.2007030299
- Paul, B. M., & Vanden Heuvel, G. B. (2014). Kidney: polycystic kidney disease. Wiley Interdisciplinary Reviews: Developmental Biology, 3(6), 465-487. https://doi.org/10.1002/wdev.152
- Subramanian, S., & Ahmad, T. (2022). Polycystic kidney disease of childhood. StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK537137/
- Torra, R. (2021, Dec 16). Polycystic Kidney Disease. Medscape. Retrieved on 2023, May 25 from https://emedicine.medscape.com/article/244907-overview
- Wilson, P. D. (2004). Polycystic kidney disease. New England Journal of Medicine, 350(2), 151-164. Doi: 10.1056/NEJMra022161